
Molecular dynamics simulation of α-unsubstituted oligo-thiophenes: dependence of their high-temperature liquid-crystalline phase behaviour on molecular length†

Abstract
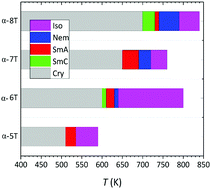
Despite that α-unsubstituted oligo-thiophenes (α-nTs) have been synthesized a long time ago and studied for many years, their phase behavior (which governs electronic performance, and thus their application in thin-film transistors) is still not fully understood. Here, we employ molecular dynamics simulations to study the high-temperature phase behaviour of α-nTs with n = 5–8 as a function of their molecular length using a recently proposed fully-flexible, united-atom model. We follow a methodology already developed for simulating the liquid-crystalline behaviour of α-sexithiophene, which we extend here to three other members of the family of α-nTs characterized by n = 5 (α-quinquethiophene), n = 7 (α-septithiophene) and n = 8 (α-octamer). Upon cooling fully pre-equilibrated bulk structures of these molecules from their amorphous, isotropic (Iso) phase at a high enough temperature down to lower temperatures, successive spontaneous phase transitions are observed leading to liquid crystalline phases. For n > 5, we observe first the formation of a nematic (Nem) phase and then the formation of two different types of smectic (Sm) phases. For n = 5, on the other hand, only one type of smectic phase is observed (no Nem was detected). We find that the type of the Sm phase formed is determined by the parity of the molecule: odd-numbered α-nTs display an orthogonal smectic A (SmA) phase whereas even-numbered α-nTs display both SmA and smectic C (SmC) phases, demonstrating a unique odd–even effect for these systems. In the SmC phase, the direction of the tilt angle is uniform, characteristic of a synclinic structure. The odd–even effect is also evident in the simulated crystalline phases of the studied α-nTs but also in other properties such as the density and the tilt angle which are found to alternate between high and low values for even- and odd-numbered α-nTs, respectively. It is interpreted here in terms of intra-molecular configurational changes taking place at the phase transition points.
 Please wait while we load your content...
Please wait while we load your content...

