
A 2-D π–π dimer model system to investigate structure-charge transfer relationships in rubrene†

Abstract
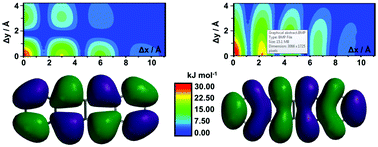
Rubrene (5,6,11,12-tetraphenyltetracene) is undoubtedly one of the best performing organic charge transfer mediating materials, with experimentally determined mobilities up to 40 cm2 V−1 s−1. Consequently, there has been increasing interest by means of crystal engineering in trying to generate rubrene-based materials with analogous or even superior conducting properties. Often, experimental measurements are carried out in thin film architectures of these materials, where measured properties can be detrimentally impacted by device manufacture rather than intrinsic charge transfer properties of the material. The latter results in discarding potential good performers. To address these concerns, we report a two-dimensional model system that will allow researchers to predict charge transfer properties of their materials solely requiring the coordinates of the π–π stacking motifs. We envisaged this study to be of significant interest to the increasingly large community of materials scientists devoted to the realisation of improved organic charge mediating materials and particularly to those engaged in exploiting rubrene-based architectures.
 Please wait while we load your content...
Please wait while we load your content...

