
Atomic-level active sites of efficient imidazolate framework-derived nickel catalysts for CO2 reduction†

Abstract
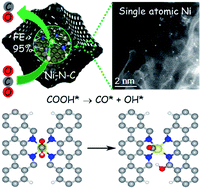
Nickel and nitrogen co-doped carbon (Ni–N–C) has emerged as a promising catalyst for the CO2 reduction reaction (CO2RR); however, the chemical nature of its active sites has remained elusive. Herein, we report the exploration of the reactivity and active sites of Ni–N–C for the CO2RR. Single atom Ni coordinated with N confined in a carbon matrix was prepared through thermal activation of chemically Ni-doped zeolitic imidazolate frameworks (ZIFs) and directly visualized by aberration-corrected scanning transmission electron microscopy. Electrochemical results show the enhanced intrinsic reactivity and selectivity of Ni–N sites for the reduction of CO2 to CO, delivering a maximum CO faradaic efficiency of 96% at a low overpotential of 570 mV. Density functional theory (DFT) calculations predict that the edge-located Ni–N2+2 sites with dangling bond-containing carbon atoms are the active sites facilitating the dissociation of the C–O bond of the *COOH intermediate, while bulk-hosted Ni–N4 is kinetically inactive. Furthermore, the high capability of edge-located Ni–N4 being able to thermodynamically suppress the competitive hydrogen evolution is also explained. The proposal of edge-hosed Ni–N2+2 sites provides new insight into designing high-efficiency Ni–N–C for CO2 reduction.
 Please wait while we load your content...
Please wait while we load your content...

