
Assessing structure and stability of polymer/lithium-metal interfaces from first-principles calculations†

Abstract
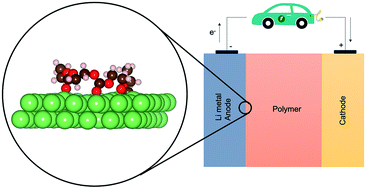
Solid polymer electrolytes (SPEs) are promising candidates for Li metal battery applications, but the interface between these two categories of materials has so far been studied only to a limited degree. A better understanding of interfacial phenomena, primarily polymer degradation, is essential for improving battery performance. The aim of this study is to get insights into atomistic surface interaction and the early stages of solid electrolyte interphase formation between ionically conductive SPE host polymers and the Li metal electrode. A range of SPE candidates are studied, representative of major host material classes: polyethers, polyalcohols, polyesters, polycarbonates, polyamines and polynitriles. Density functional theory (DFT) calculations are carried out to study the stability and the electronic structure of such polymer/Li interfaces. The adsorption energies indicated a stronger adhesion to Li metal of polymers with ester/carbonate and nitrile functional groups. Together with a higher charge redistribution, a higher reactivity of these polymers is predicted as compared to the other electrolyte hosts. Products such as alkoxides and CO are obtained from the degradation of ester- and carbonate-based polymers by AIMD simulations, in agreement with experimental studies. Analogous to low-molecular-weight organic carbonates, decomposition pathways through Ccarbonyl–Oethereal and Cethereal–Oethereal bond cleavage can be assumed, with carbonate-containing fragments being thermodynamically favorable.
 Please wait while we load your content...
Please wait while we load your content...

