
Enhanced Li-ion dynamics in trivalently doped lithium phosphidosilicate Li2SiP2: a candidate material as a solid Li electrolyte†

Abstract
Oxide and sulphide solid electrolyte materials have enjoyed significant interest in the solid-state battery community. Phosphide materials however are relatively unexplored despite the potential for being high lithium containing systems. This work reports on the phosphidosilicate system Li2SiP2, one of many systems in the Li–Si–P phase diagram. The phosphidosilicates display complex structures and very large unit cells, which present challenges for ab initio simulations. We present the first computational report on the theoretical ionic conductivity and related diffusion mechanisms of the material Li2SiP2, selected due to it's unusual supertetrahedral framework which is a recurrent motif amongst the phosphidosilicates. Group 13 dopants have also been introduced into Li2SiP2 showing preference for the silicon site over the lithium site, with 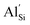 doping showing extremely low defect incorporation energies of 0.05 eV, with no increase in defect energy up to concentrations of 10%
doping showing extremely low defect incorporation energies of 0.05 eV, with no increase in defect energy up to concentrations of 10% 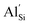 . Furthermore, clustering of
. Furthermore, clustering of 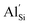 has been found to be unfavourable, in line with trends seen in oxide zeolite structures. Ab initio molecular dynamics (AIMD) simulations indicate high ionic conductivity in pure Li2SiP2 of up to 3.19 × 10−1 S cm−1 at 700 K. Doping with 10%
has been found to be unfavourable, in line with trends seen in oxide zeolite structures. Ab initio molecular dynamics (AIMD) simulations indicate high ionic conductivity in pure Li2SiP2 of up to 3.19 × 10−1 S cm−1 at 700 K. Doping with 10% 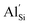 and associated
and associated 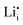 compensating defects leads to higher ionic conductivities at lower temperatures when compared to pure Li2SiP2. The activation energies to lithium diffusion were found to be low at 0.30 eV and 0.24 eV for pure and 10%
compensating defects leads to higher ionic conductivities at lower temperatures when compared to pure Li2SiP2. The activation energies to lithium diffusion were found to be low at 0.30 eV and 0.24 eV for pure and 10% 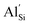 doped Li2SiP2 respectively, in line with previous experimental observations of pure Li2SiP2. Multiple lithium migration pathways have also been extracted, with some mechanisms displaying activation energies as low as 0.05 eV. Furthermore, our calculated intercalation voltages suggest that these materials are stable against lithium metal and therefore could be very attractive in stabilising the electrode/electrolyte interface.
doped Li2SiP2 respectively, in line with previous experimental observations of pure Li2SiP2. Multiple lithium migration pathways have also been extracted, with some mechanisms displaying activation energies as low as 0.05 eV. Furthermore, our calculated intercalation voltages suggest that these materials are stable against lithium metal and therefore could be very attractive in stabilising the electrode/electrolyte interface.

 Please wait while we load your content...
Please wait while we load your content...

