
CO2 reduction with protons and electrons at a boron-based reaction center†

Abstract
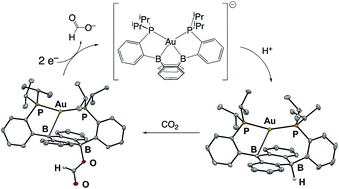
Borohydrides are widely used reducing agents in chemical synthesis and have emerging energy applications as hydrogen storage materials and reagents for the reduction of CO2. Unfortunately, the high energy cost associated with the multistep preparation of borohydrides starting from alkali metals precludes large scale implementation of these latter uses. One potential solution to this issue is the direct synthesis of borohydrides from the protonation of reduced boron compounds. We herein report reactions of the redox series [Au(B2P2)]n (n = +1, 0, −1) (B2P2, 9,10-bis(2-(diisopropylphosphino)phenyl)-9,10-dihydroboranthrene) and their conversion into corresponding mono- and diborohydride complexes. Crucially, the monoborohydride can be accessed via protonation of [Au(B2P2)]−, a masked borane dianion equivalent accessible at relatively mild potentials (−2.05 V vs. Fc/Fc+). This species reduces CO2 to produce the corresponding formate complex. Cleavage of the formate complex can be achieved by reduction (ca. −1.7 V vs. Fc/Fc+) or by the addition of electrophiles including H+. Additionally, direct reaction of [Au(B2P2)]− with CO2 results in reductive disproportion to release CO and generate a carbonate complex. Together, these reactions constitute a synthetic cycle for CO2 reduction at a boron-based reaction center that proceeds through a B–H unit generated via protonation of a reduced borane with weak organic acids.
- This article is part of the themed collection: Spotlighting main group elements in polynuclear complexes
 Please wait while we load your content...
Please wait while we load your content...

