
Thermodynamic and kinetic studies of H2 and N2 binding to bimetallic nickel-group 13 complexes and neutron structure of a Ni(η2-H2) adduct†
 ‡abe
Samantha A.
Burgess,
c
Konstantinos D.
Vogiatzis,
abf
Jingyun
Ye,
ab
John C.
Linehan,
c
Aaron M.
Appel,
c
Christina
Hoffmann,
d
Xiaoping
Wang,
d
Connie C.
Lu
*a
‡abe
Samantha A.
Burgess,
c
Konstantinos D.
Vogiatzis,
abf
Jingyun
Ye,
ab
John C.
Linehan,
c
Aaron M.
Appel,
c
Christina
Hoffmann,
d
Xiaoping
Wang,
d
Connie C.
Lu
*a
Abstract
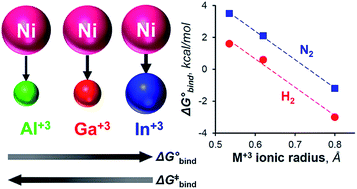
Understanding H2 binding and activation is important in the context of designing transition metal catalysts for many processes, including hydrogenation and the interconversion of H2 with protons and electrons. This work reports the first thermodynamic and kinetic H2 binding studies for an isostructural series of first-row metal complexes: NiML, where M = Al (1), Ga (2), and In (3), and L = [N(o-(NCH2PiPr2)C6H4)3]3−. Thermodynamic free energies (ΔG°) and free energies of activation (ΔG‡) for binding equilibria were obtained via variable-temperature 31P NMR studies and lineshape analysis. The supporting metal exerts a large influence on the thermodynamic favorability of both H2 and N2 binding to Ni, with ΔG° values for H2 binding found to span nearly the entire range of previous reports. The non-classical H2 adduct, (η2-H2)NiInL (3-H2), was structurally characterized by single-crystal neutron diffraction—the first such study for a Ni(η2-H2) complex or any d10 M(η2-H2) complex. UV-Vis studies and TD-DFT calculations identified specific electronic structure perturbations of the supporting metal which poise NiML complexes for small-molecule binding. ETS-NOCV calculations indicate that H2 binding primarily occurs via H–H σ-donation to the Ni 4pz-based LUMO, which is proposed to become energetically accessible as the Ni(0)→M(III) dative interaction increases for the larger M(III) ions. Linear free-energy relationships are discussed, with the activation barrier for H2 binding (ΔG‡) found to decrease proportionally for more thermodynamically favorable equilibria. The ΔG° values for H2 and N2 binding to NiML complexes were also found to be more exergonic for the larger M(III) ions.
- This article is part of the themed collections: Spotlighting main group elements in polynuclear complexes and Most popular 2019-2020 inorganic, main group and crystal engineering chemistry articles
 Please wait while we load your content...
Please wait while we load your content...

