
Mapping free energy regimes in electrocatalytic reductions to screen transition metal-based catalysts†

Abstract
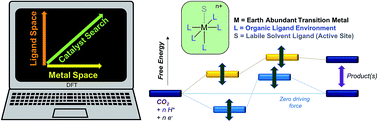
The free energy landscape of catalytic intermediates in the two-electron reduction of proton donors and/or CO2 to H2, CO and HCO2− is mapped with density functional theory to screen catalyst candidates from a library of different transition metals and ligands. The goal is to minimize the free energy corrugations between reactants, catalytic intermediates and each desired product, simultaneously screening against intermediates with low free energy that would be traps, and against necessary intermediates with high free energy. Catalysts are initially screened for those with: (a) standard state free energy of the metal hydride intermediate ergoneutral with HCO2−, which is the lowest energy product with weak proton donors, and (b) standard free energy of the metal carbonyl intermediate sufficiently high to avoid trapping. The design method is tested on a diverse range of ligands including cyclopentadienyl, polypyridyl, amino, phosphino and carbonyl ligands, around three earth-abundant d6 transition metal ions, Mn(I), Fe(II) and Co(III), using the BP86 density functional, the double-zeta 6-31+G* basis, LANL2DZ effective core potential on the metals and SMD polarizable continuum model for acetonitrile as solvent, which have previously provided chemically accurate values of several redox potentials, pKa's and ligand exchange equilibria for transition metal complexes. Among the 36 complexes screened, an Fe(II) center ligated to two bipyridines and a pyridine with a solvent-bound sixth coordination site for hydride formation from phenol as the proton donor is identified as a promising candidate for ergoneutral hydride formation without trapping by CO. The redox-active bipyridine ligands are predicted to provide near ergoneutral sites for accumulating the two electrons needed to form the hydride. To test the predictions, an Fe(II) complex was prepared with the desired ligand environment using a pentadentate ligand to prevent ligand exchange. The synthesized complex was indeed found to be active towards electrocatalytic proton reduction as well as CO2 reduction at the predicted redox potentials with no trapping by CO. However, contrary to the in silico predictions, we found electrochemical evidence of CO2 binding after the first reduction leading to CO production. Mapping the free energies of key catalytic intermediates such as the metal hydride and metal carbonyl species by using density functional theory (DFT) serves as a first step in catalyst screening spanning large libraries of metals and ligands. In order to screen against all the intermediates in the catalytic pathway, such as reduced metal-bound CO2 intermediates, further refinement and validation of the DFT methods are needed.
 Please wait while we load your content...
Please wait while we load your content...

