
Towards large scale hybrid QM/MM dynamics of complex systems with advanced point dipole polarizable embeddings†
 *a
Gérardo A.
Cisneros,
d
Filippo
Lipparini,
f
Benedetta
Mennucci
f
and
Jean-Philip
Piquemal
*agh
*a
Gérardo A.
Cisneros,
d
Filippo
Lipparini,
f
Benedetta
Mennucci
f
and
Jean-Philip
Piquemal
*agh
Abstract
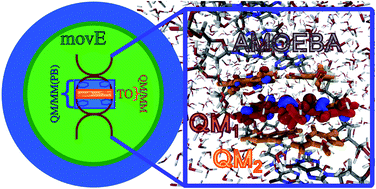
In this work, we present a general route to hybrid Quantum Mechanics/Molecular Mechanics (QM/MM) Molecular Dynamics for complex systems using a polarizable embedding. We extend the capabilities of our hybrid framework, combining the Gaussian and Tinker/Tinker-HP packages in the context of the AMOEBA polarizable force field to treat large (bio)systems where the QM and the MM subsystems are covalently bound, adopting pseudopotentials at the boundaries between the two regions. We discuss in detail the implementation and demonstrate the global energy conservation of our QM/MM Born–Oppenheimer molecular dynamics approach using Density Functional Theory. Finally, the approach is assessed on the electronic absorption properties of a 16 500 atom complex encompassing an organic dye embedded in a DNA matrix in solution, extending the hybrid method to a time-dependent Density Functional Theory approach. The results obtained comparing different partitions between the quantum and the classical subsystems also suggest that large QM portions are not necessary if accurate polarizable force fields are used in a variational formulation of the embedding, properly including the QM/MM mutual polarization.
 Please wait while we load your content...
Please wait while we load your content...

