
Bayesian semi-supervised learning for uncertainty-calibrated prediction of molecular properties and active learning
 *a
*a
Abstract
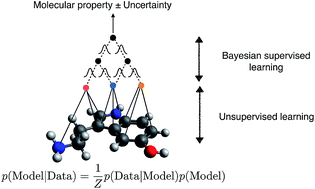
Predicting bioactivity and physical properties of small molecules is a central challenge in drug discovery. Deep learning is becoming the method of choice but studies to date focus on mean accuracy as the main metric. However, to replace costly and mission-critical experiments by models, a high mean accuracy is not enough: outliers can derail a discovery campaign, thus models need to reliably predict when it will fail, even when the training data is biased; experiments are expensive, thus models need to be data-efficient and suggest informative training sets using active learning. We show that uncertainty quantification and active learning can be achieved by Bayesian semi-supervised graph convolutional neural networks. The Bayesian approach estimates uncertainty in a statistically principled way through sampling from the posterior distribution. Semi-supervised learning disentangles representation learning and regression, keeping uncertainty estimates accurate in the low data limit and allowing the model to start active learning from a small initial pool of training data. Our study highlights the promise of Bayesian deep learning for chemistry.
 Please wait while we load your content...
Please wait while we load your content...

