
Learning continuous and data-driven molecular descriptors by translating equivalent chemical representations†

Abstract
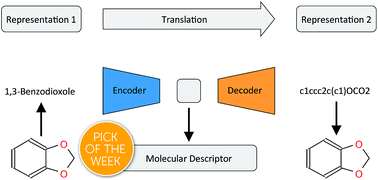
There has been a recent surge of interest in using machine learning across chemical space in order to predict properties of molecules or design molecules and materials with the desired properties. Most of this work relies on defining clever feature representations, in which the chemical graph structure is encoded in a uniform way such that predictions across chemical space can be made. In this work, we propose to exploit the powerful ability of deep neural networks to learn a feature representation from low-level encodings of a huge corpus of chemical structures. Our model borrows ideas from neural machine translation: it translates between two semantically equivalent but syntactically different representations of molecular structures, compressing the meaningful information both representations have in common in a low-dimensional representation vector. Once the model is trained, this representation can be extracted for any new molecule and utilized as a descriptor. In fair benchmarks with respect to various human-engineered molecular fingerprints and graph-convolution models, our method shows competitive performance in modelling quantitative structure–activity relationships in all analysed datasets. Additionally, we show that our descriptor significantly outperforms all baseline molecular fingerprints in two ligand-based virtual screening tasks. Overall, our descriptors show the most consistent performances in all experiments. The continuity of the descriptor space and the existence of the decoder that permits deducing a chemical structure from an embedding vector allow for exploration of the space and open up new opportunities for compound optimization and idea generation.
 Please wait while we load your content...
Please wait while we load your content...

