
Reduction of CO2 by a masked two-coordinate cobalt(i) complex and characterization of a proposed oxodicobalt(ii) intermediate†‡
 §ag
Bhaskar
Mondal,
a
Kyle M.
Lancaster,
d
Jason
Shearer,
e
William W.
Brennessel,
b
Shengfa
Ye
*f
and
Patrick L.
Holland
*c
§ag
Bhaskar
Mondal,
a
Kyle M.
Lancaster,
d
Jason
Shearer,
e
William W.
Brennessel,
b
Shengfa
Ye
*f
and
Patrick L.
Holland
*c
Abstract
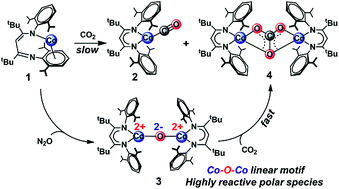
Fixation and chemical reduction of CO2 are important for utilization of this abundant resource, and understanding the detailed mechanism of C–O cleavage is needed for rational development of CO2 reduction methods. Here, we describe a detailed analysis of the mechanism of the reaction of a masked two-coordinate cobalt(I) complex, LtBuCo (where LtBu = 2,2,6,6-tetramethyl-3,5-bis[(2,6-diisopropylphenyl)imino]hept-4-yl), with CO2, which yields two products of C–O cleavage, the cobalt(I) monocarbonyl complex LtBuCo(CO) and the dicobalt(II) carbonate complex (LtBuCo)2(μ-CO3). Kinetic studies and computations show that the κN,η6-arene isomer of LtBuCo rearranges to the κ2N,N′ binding mode prior to binding of CO2, which contrasts with the mechanism of binding of other substrates to LtBuCo. Density functional theory (DFT) studies show that the only low-energy pathways for cleavage of CO2 proceed through bimetallic mechanisms, and DFT and highly correlated domain-based local pair natural orbital coupled cluster (DLPNO-CCSD(T)) calculations reveal the cooperative effects of the two metal centers during facile C–O bond rupture. A plausible intermediate in the reaction of CO2 with LtBuCo is the oxodicobalt(II) complex LtBuCoOCoLtBu, which has been independently synthesized through the reaction of LtBuCo with N2O. The rapid reaction of LtBuCoOCoLtBu with CO2 to form the carbonate product indicates that the oxo species is kinetically competent to be an intermediate during CO2 cleavage by LtBuCo. LtBuCoOCoLtBu is a novel example of a thoroughly characterized molecular cobalt–oxo complex where the cobalt ions are clearly in the +2 oxidation state. Its nucleophilic reactivity is a consequence of high charge localization on the μ-oxo ligand between two antiferromagnetically coupled high-spin cobalt(II) centers, as characterized by DFT and multireference complete active space self-consistent field (CASSCF) calculations.
- This article is part of the themed collection: Celebrating the Chemical Sciences in India - Leaders in the Field Symposium 2020
 Please wait while we load your content...
Please wait while we load your content...

