
Effects of cooling rate on structural relaxation in amorphous drugs: elastically collective nonlinear langevin equation theory and machine learning study
 *abc
Katsunori
Wakabayashi,
c
*abc
Katsunori
Wakabayashi,
c
Abstract
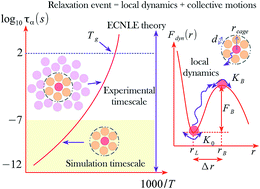
Theoretical approaches are formulated to investigate the molecular mobility under various cooling rates of amorphous drugs. We describe the structural relaxation of a tagged molecule as a coupled process of cage-scale dynamics and collective molecular rearrangement beyond the first coordination shell. The coupling between local and non-local dynamics behaves distinctly in different substances. Theoretical calculations for the structural relaxation time, glass transition temperature, and dynamic fragility are carried out over twenty-two amorphous drugs and polymers. Numerical results have a quantitatively good accordance with experimental data and the extracted physical quantities using the Vogel–Fulcher–Tammann fit function and machine learning. The machine learning method reveals the linear relation between the glass transition temperature and the melting point, which is a key factor for pharmaceutical solubility. Our predictive approaches are reliable tools for developing drug formulations.
- This article is part of the themed collection: Machine learning and artificial neural networks in chemistry
 Please wait while we load your content...
Please wait while we load your content...

