
Adequate prediction for inhibitor affinity of Aβ40 protofibril using the linear interaction energy method†

Abstract
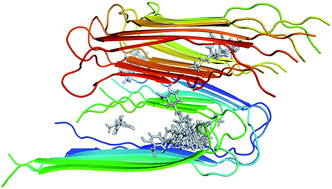
The search for efficient inhibitors targeting Aβ oligomers and fibrils is an important issue in Alzheimer's disease treatment. As a consequence, an accurate and computationally cheap approach to estimate the binding affinity for many ligands interacting with Aβ peptides is very important. Here, the calculated binding free energies of 30 ligands interacting with 12Aβ11–40 peptides using the linear interaction energy (LIE) approach are found to be in good correlation with experimental data (R = 0.79). The binding affinities of these complexes are also calculated by using free energy perturbation (FEP) and molecular mechanic/Poisson–Boltzmann surface area (MM/PBSA) methods. The time-consuming FEP method provides results with similar correlation (R = 0.72), whereas MM/PBSA calculations show very low correlation with experimental data (R = 0.27). In all complexes, van der Waals interactions contribute much more than electrostatic interactions. The LIE model, which is much less time-consuming than both the FEP and MM/PBSA methods, opens the door to accurate and rapid affinity prediction of ligands with Aβ peptides and the design of new ligands.
 Please wait while we load your content...
Please wait while we load your content...

