
The poly(propylene oxide-co-ethylene oxide) gradient is controlled by the polymerization method: determination of reactivity ratios by direct comparison of different copolymerization models†
 *a
*a
Abstract
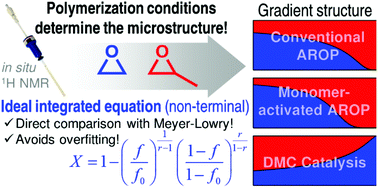
An investigation of the highly relevant copolymerization of ethylene oxide (EO) and propylene oxide (PO) by in situ1H NMR spectroscopy shows striking differences in the copolymerization kinetics, depending on the polymerization method. Examination of the EO/PO copolymerization kinetics using iBu3Al for the monomer-activated anionic ring opening polymerization (AROP) confirmed a strong monomer gradient of the microstructure (rEO = 6.4, rPO = 0.16) in contrast to the known weak gradient in the conventional AROP (rEO = 2.8, rPO = 0.25). The first study via in situ1H-NMR kinetics of the copolymerization of PO and EO under heterogeneous double metal cyanide (DMC) catalysis, a method that produces megatons of polyether polyols in industry, revealed a reversal of the monomer gradient (rEO = 0.42, rPO = 2.4). Thus, the copolymer microstructure of these polyether copolymers can be specifically adjusted depending on the choice of the polymerization method. The in situ1H NMR kinetics data were fitted to both the non-terminal and terminal copolymerization models. To directly compare the fits of both models, a new copolymerization equation for the non-terminal model was derived by solving the Skeist-relation analytically in analogy to the Meyer–Lowry equation. This newly derived equation allows the direct comparison of both models without transformation of the in situ data for the first time. Thus, the ideal integrated equation can help to recognize overfitting of copolymerization data. Furthermore, the equation was proven to give good estimates for reactivity ratios of ideal copolymerizations, even when systematic errors were introduced. Additionally, the obtained reactivity ratios were used to perform kinetic Monte Carlo simulations to visualize the EO/PO copolymer microstructure and to determine the nature of the terminal monomer unit.
 Please wait while we load your content...
Please wait while we load your content...

