
Temperature-induced molecular reorganization on Au(111) driven by oligomeric defects†
 ‡a
G.
Galeotti,
‡ab
M.
Simenas,
c
M. C.
Gallagher,
*d
E.
Hamzehpoor,
e
O.
MacLean,
a
D.
Dettmann,
b
G.
Contini,
bf
M.
Ebrahimi,
§*a
D. F.
Perepichka
*e
and
F.
Rosei
*a
‡a
G.
Galeotti,
‡ab
M.
Simenas,
c
M. C.
Gallagher,
*d
E.
Hamzehpoor,
e
O.
MacLean,
a
D.
Dettmann,
b
G.
Contini,
bf
M.
Ebrahimi,
§*a
D. F.
Perepichka
*e
and
F.
Rosei
*a
Abstract
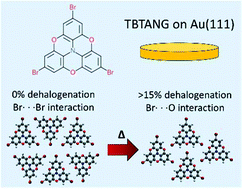
The formation of ordered molecular structures on surfaces is determined by the balance between molecule–molecule and molecule–substrate interactions. Whether the aggregation process is guided by non-covalent forces or on-surface reactions, a deeper understanding of these interactions is pivotal to formulating a priori predictions of the final structural features and the development of bottom-up fabrication protocols. Theoretical models of molecular systems corroborate the information gathered through experimental observations and help explain the thermodynamic factors that underpin on-surface phase transitions. Here, we report a scanning tunneling microscopy investigation of a tribromo-substituted heterotriangulene on the Au(111) surface, which initially forms an extended close-packed ordered structure stabilized by Br⋯Br halogen bonds when deposited at room temperature. X-ray photoelectron spectroscopy reveals that annealing the self-assembled layer induces a fraction of the molecular precursors to partially dehalogenate that in turn leads to the formation of a less stable Br⋯O non-covalent network which coexists with the short oligomers. Density functional theory (DFT) and Monte Carlo (MC) simulations illustrate how dimer moieties act as defects whose steric hindrance prevents the retention of the more stable configuration. A small number of dimers is sufficient to drive the molecular reorganization into a lower cohesive energy phase. Our study shows the importance of a combined DFT – MC approach to understand the evolution of molecular systems on substrates.
- This article is part of the themed collection: Nanoscale 10th Anniversary Special Issue
 Please wait while we load your content...
Please wait while we load your content...

