
Interface dipoles of Ir(ppy)3 on Cu(111)†

Abstract
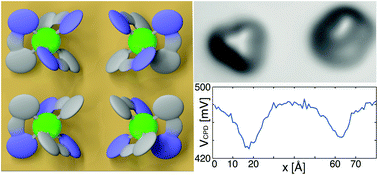
The interplay of adsorption geometry and interface dipoles of the transition-metal complex Ir(ppy)3 on Cu(111) was studied using low-temperature scanning probe microscopy and density-functional-theory calculations. We find that the orientation of the molecule's intrinsic dipole moment with respect to the surface has a strong influence on the total energy of the different configurations, where the most stable one has the molecular dipole moment pointing out of the surface plane along the surface normal. Adsorption-induced redistribution of charges results in an additional dipole moment that also points out of the surface plane for all configurations. Submolecularly resolved maps of the resulting local contact potential difference suggest that any in-plane dipole moment is very effectively screened.
 Please wait while we load your content...
Please wait while we load your content...

