
Crystal structures of N6-modified-amino acid related nucleobase analogs (II): hybrid adenine-β-alanine and adenine-GABA molecules†

Abstract
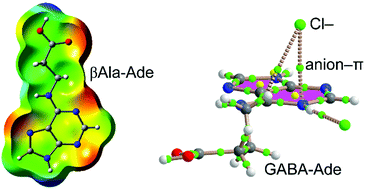
In this manuscript we report the synthesis and X-ray characterization of four N6-amino acid/peptide-adenine-derivatives: N6-βAlaAde·1.5H2O (1) and N6-GABAAde·2H2O (2) and their corresponding protonated forms N6-βAlaAde·HCl (3) and N6-GABAAde·HCl (4). In (1) with a neutral adenine ring, the protonated carboxylate interacts with the N(7) and N(6)H of the neighbouring molecule. The hydrogen bond N9–H⋯N(3) and the hydrogen bonds between the water molecules are responsible for the planar and parallel disposition of the adenine rings. In (2), two different molecules are present in the crystal structure: (a) a cationic unit in which the N(7)H tautomeric adenine is protonated at N(3) and the carboxylic group interacts with N(6B)–H and N(7B) of the adjacent molecule; (b) an anionic unit, which presents the adenine ring in the N(9)H tautomeric form, where the carboxylate interacts with the N(7A)H and N(6A)H of the neighbouring adenine. In the hydrochloride form of N6-βAlaAde (compound 3) the amino acid chain with the carboxylic acid is almost orthogonal to the ring plane and exhibits protonation at N(3) of the adenine. On the other hand, in compound (4), the side chain is arranged parallel to the ring and anion (Cl−)–π interactions are responsible for a parallel ordering of the final solid state architecture. We have studied the noncovalent interactions observed in the solid state architecture energetically using DFT calculations and rationalized the interactions using Molecular Electrostatic Potential surfaces and Bader's theory of “Atoms-in-Molecules”. The main purpose of this study is to explore the competition between homodimer formation by the Hoogsteen site of the adeninium cation, and self-association of the carboxylic group or through the interaction of the carboxylic group with the adeninium cation by X-ray crystallography.
 Please wait while we load your content...
Please wait while we load your content...

