
Surpassing 10 000 identified and quantified proteins in a single run by optimizing current LC-MS instrumentation and data analysis strategy†
 ‡a
Tejas
Gandhi,
‡a
Lynn
Verbeke,
a
Oliver M.
Bernhardt,
a
Roland
Bruderer
a
and
Lukas
Reiter
*a
‡a
Tejas
Gandhi,
‡a
Lynn
Verbeke,
a
Oliver M.
Bernhardt,
a
Roland
Bruderer
a
and
Lukas
Reiter
*a
Abstract
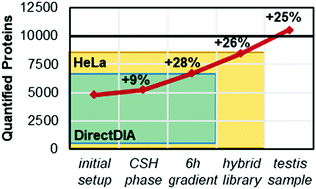
Comprehensive proteome quantification is crucial for a better understanding of underlying mechanisms of diseases. Liquid chromatography mass spectrometry (LC-MS) has become the method of choice for comprehensive proteome quantification due to its power and versatility. Even though great advances have been made in recent years, full proteome coverage for complex samples remains challenging due to the high dynamic range of protein expression. Additionally, when studying disease regulatory proteins, biomarkers or potential drug targets are often low abundant, such as for instance kinases and transcription factors. Here, we show that with improvements in chromatography and data analysis the single shot proteome coverage can go beyond 10 000 proteins in human tissue. In a testis cancer study, we quantified 11 200 proteins using data independent acquisition (DIA). This depth was achieved with a false discovery rate of 1% which was experimentally validated using a two species test. We introduce the concept of hybrid libraries which combines the strength of direct searching of DIA data as well as the use of large project-specific or published DDA data sets. Remarkably deep proteome coverage is possible using hybrid libraries without the additional burden of creating a project-specific library. Within the testis cancer set, we found a large proportion of proteins in an altered expression (in total: 3351; 1453 increased in cancer). Many of these proteins could be linked to the hallmarks of cancer. For example, the complement system was downregulated which helps to evade the immune response and chromosomal replication was upregulated indicating a dysregulated cell cycle.
 Please wait while we load your content...
Please wait while we load your content...