
Structural and chemical properties of half-sandwich rhodium complexes supported by the bis(2-pyridyl)methane ligand†

Abstract
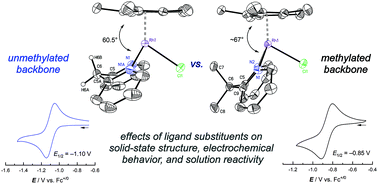
[Cp*Rh] complexes (Cp* = pentamethylcyclopentadienyl) supported by bidentate chelating ligands are a useful class of compounds for studies of redox chemistry and catalysis. Here, we show that the bis(2-pyridyl)methane ligand, also known as dipyridylmethane or dpma, can support [Cp*Rh] complexes in the formally +III and +II rhodium oxidation states. Specifically, two new rhodium complexes ([Cp*Rh(dpma)(L)]n+, L = Cl−, CH3CN) have been isolated and structurally characterized, and the properties of the complexes have been compared with those of [Cp*Rh] complexes bearing the related dimethyldipyridylmethane (Me2dpma) ligand. Complex [Cp*Rh(dpma)(NCCH3)]2+ displays a quasireversible rhodium(III/II) reduction by cyclic voltammetry; related electron paramagnetic resonance (EPR) spectroscopic studies confirm access to the unusual rhodium(II) oxidation state. Further reduction to the formally rhodium(I) oxidation state, however, is followed by deprotonation of dpma, as observed in electrochemical studies and chemical reduction experiments. This reactivity can be understood to occur as a consequence of the presence of doubly benzylic protons in the dpma ligand, since use of the analogous Me2dpma enables reduction to rhodium(I) without involvement of ligand deprotonation. These findings highlight the important role of the ligand backbone substitution pattern in influencing the stability of highly-reduced complexes, a key class of metal species for study of electron and proton management in catalysis.
 Please wait while we load your content...
Please wait while we load your content...

