
Four-component relativistic 31P NMR calculations for trans-platinum(ii) complexes: importance of the solvent and dynamics in spectral simulations†
 a
Heike
Fliegl,
b
Michele
Cascella,
b
Stanislav
Komorovsky,
cd
Adoración G.
Quiroga
e
and
Marcel
Swart
*af
a
Heike
Fliegl,
b
Michele
Cascella,
b
Stanislav
Komorovsky,
cd
Adoración G.
Quiroga
e
and
Marcel
Swart
*af
Abstract
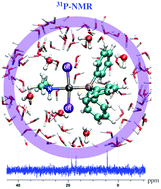
We report a combined experimental–theoretical study on the 31P NMR chemical shift for a number of trans-platinum(II) complexes. Validity and reliability of the 31P NMR chemical shift calculations are examined by comparing with the experimental data. A successful computational protocol for the accurate prediction of the 31P NMR chemical shifts was established for trans-[PtCl2(dma)PPh3] (dma = dimethylamine) complexes. The reliability of the computed values is shown to be critically dependent on the level of relativistic effects (two-component vs. four component), choice of density functionals, dynamical averaging, and solvation effects. Snapshots obtained from ab initio molecular dynamics simulations were used to identify those solvent molecules which show the largest interactions with the platinum complex, through inspection by using the non-covalent interaction program. We observe satisfactory accuracy from the full four-component matrix Dirac–Kohn–Sham method (mDKS) based on the Dirac–Coulomb Hamiltonian, in conjunction with the KT2 density functional, and dynamical averaging with explicit solvent molecules.
 Please wait while we load your content...
Please wait while we load your content...

