
Pathways of selective catalytic CO2 two-step reduction on di-iron, di-cobalt and iron-cobalt disulfide carbonyls – an electronic structure study†

Abstract
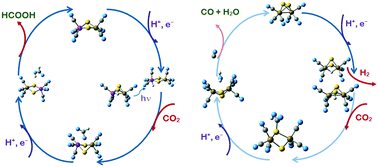
The hexacarbonyl clusters of bimetal disulfides [2Fe-2S], [Fe, Co-2S] and [2Co-2S] with cyclic or rhombic cores of sulphur-bridged metal cations in the divalent state provide an energetically favourable path for the two-step reduction of CO2, as determined by density functional theory calculations. The elementary steps of the reduction to formic acid, HCOOH, or CO + H2O are traced by transition state theory. The umbrella-shaped three-carbonyl ligands at each cation induce a high electron density in the [2M-2S] core and they also impose stereo-selectivity by limiting the mobility of adsorbed reactants and intermediates. The hexacarbonyl complexes have high proton affinities, 700–850 kJ mol−1. Inclusion of cobalt ensures high proton–electron affinities (H+, e−) of 250–300 kJ mol−1 and cobalt is able to directly coordinate and activate hydrogen. In the [CoFeS2(H+, e−)](CO)6 complexes charge separation is observed, the proton being attached to sulphur, whereas the electron is delocalised at the metal cation centres. The parent hexacarbonyl complexes and their reduced forms after accepting the proton–electron couple have intense excitation lines in the visible spectrum. While the preferred position for H-binding is at a sulphur atom, visible light excitations can promote the sulphur-bonded proton to cobalt-bonded active hydrogen. The inclusion of cobalt thus provides a selective, low-energy path of two proton–electron reduction to formic acid. Cobalt centres transfer the activated hydrogen to the carbon dioxide molecule so as to form a HCO2˙ intermediate and this is the rate-determining step. The di-iron disulfide [2Fe-2S] complexes act in a different way: hydrogen is either fixed at a sulphur centre or shared by the two iron cations and it is transferred preferably to the oxygen end of a CO2 molecule to form a COOH˙ intermediate. The path to formic acid is predetermined by cobalt-bonded hydrogen transfer to the carbon centre of CO2 in the first reduction step. Catalyst decay paths are considered and high stability of the cobalt-containing complexes is revealed.
 Please wait while we load your content...
Please wait while we load your content...

