
Beyond the Coulson–Fischer point: characterizing single excitation CI and TDDFT for excited states in single bond dissociations†

Abstract
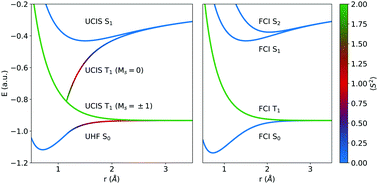
Linear response time dependent density functional theory (TDDFT), which builds upon configuration interaction singles (CIS) and TD-Hartree–Fock (TDHF), is the most widely used class of excited state quantum chemistry methods and is often employed to study photochemical processes. This paper studies the behavior of the resulting excited state potential energy surfaces beyond the Coulson–Fischer (CF) point in single bond dissociations, when the optimal reference determinant is spin-polarized. Many excited states exhibit sharp kinks at the CF point, and connect to different dissociation limits via a zone of unphysical concave curvature. In particular, the unrestricted MS = 0 lowest triplet T1 state changes character, and does not dissociate into ground state fragments. The unrestricted MS = ±1 T1 CIS states better approximate the physical dissociation limit, but their degeneracy is broken beyond the CF point for most single bond dissociations. On the other hand, the MS = ±1 T1 TDHF states reach the asymptote too soon, by merging with the ground state from the CF point onwards. Use of local exchange–correlation functionals causes MS = ±1 T1 TDDFT states to resemble their unphysical MS = 0 counterpart. The 2 orbital, 2-electron model system of minimal basis H2 is analytically treated to understand the origin of these issues, revealing that the lack of double excitations is at the root of these remarkable observations. The behavior of excited state surfaces is also numerically examined for species like H2, NH3, C2H6 and LiH in extended basis sets.
- This article is part of the themed collections: PCCP Editor’s Choice, 2020 and 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

