
Calculation of vibrationally resolved absorption spectra of acenes and pyrene†

Abstract
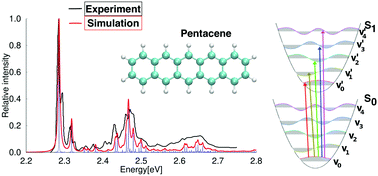
The absorption spectra of naphthalene, anthracene, pentacene and pyrene in the ultraviolet-visible (UV-Vis) range have been simulated by using an efficient real-time generating function method that combines calculated adiabatic electronic excitation energies with vibrational energies of the excited states. The vertical electronic excitation energies have been calculated at the density functional theory level using the PBE0 functional and at the second-order approximate coupled-cluster level (CC2). The absorption spectra have been calculated at the PBE0 level for the studied molecules and at the CC2 level for naphthalene. The transition probabilities between vibrationally resolved states were calculated by using the real-time generating function method employing the full Duschinsky formalism. The absorption spectrum for naphthalene calculated at the PBE0 and CC2 levels agrees well with the experimental one after the simulated spectra have been blue-shifted by 0.48 eV and 0.12 eV at the PBE0 and CC2 level, respectively. The absorption spectra for anthracene, pentacene and pyrene simulated at the PBE0 level agree well with the experimental ones when they are shifted by 0.49 eV, 0.57 eV and 0.46 eV, respectively. The strongest transitions of the main vibrational bands have been assigned.
 Please wait while we load your content...
Please wait while we load your content...

