
Extensive tests and evaluation of the CHARMM36IDPSFF force field for intrinsically disordered proteins and folded proteins†
 a
Hai-Feng
Chen
*ac
a
Hai-Feng
Chen
*ac
Abstract
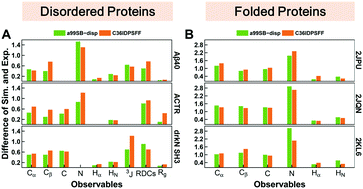
Intrinsically disordered proteins (IDPs) have received increasing attention in recent studies due to their structural heterogeneity and critical biological functions. To fully understand the structural properties and determine accurate ensembles of IDPs, molecular dynamics (MD) simulation was widely used to sample diverse conformations and reveal the structural dynamics. However, the classical state-of-the-art force fields perform well for folded proteins while being unsatisfactory for the simulations of disordered proteins reported in many previous studies. Thus, improved force fields were developed to precisely describe both folded proteins and disordered proteins. Preliminary tests show that our newly developed CHARMM36IDPSFF (C36IDPSFF) force field can well reproduce the experimental observables of several disordered proteins, but more tests on different types of proteins are needed to further evaluate the performance of C36IDPSFF. Here, we extensively simulate short peptides, disordered proteins, and fast-folding proteins as well as folded proteins, and compare the simulated results with the experimental observables. The simulation results show that C36IDPSFF could substantially reproduce the experimental observables for most of the tested proteins but some limitations are also found in the radius of gyration of large disordered proteins and the stability of fast-folding proteins. This force field will facilitate large scale studies of protein structural dynamics and functions using MD simulations.
 Please wait while we load your content...
Please wait while we load your content...

