
Multistate hybrid time-dependent density functional theory with surface hopping accurately captures ultrafast thymine photodeactivation

Abstract
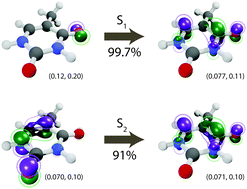
We report an efficient analytical implementation of first-order nonadiabatic derivative couplings between arbitrary Born–Oppenheimer states in the hybrid time-dependent density functional theory (TDDFT) framework using atom-centered basis functions. Our scheme is based on quadratic response theory and includes orbital relaxation terms neglected in previous approaches. Simultaneous computation of multiple derivative couplings and energy gradients enables efficient multistate nonadiabatic molecular dynamics simulations in conjunction with Tully's fewest switches surface hopping (SH) method. We benchmark the thus obtained multistate TDDFT-SH scheme by simulating ultrafast decay of UV-photoexcited thymine, for which accurate gas-phase data from ultrafast spectroscopy experiments are available. The calculations predict a fast 153 fs decay from the bright S2 to the dark S1 excited state, followed by a much slower 14 ps S1 deactivation to the ground state; statistical uncertainties were estimated using bootstrap sampling. These results agree well with the experimentally observed time constants of 100–200 fs and 5–7 ps, respectively, unlike previous multiconfigurational self-consistent field and second-order algebraic diagrammatic construction calculations. Furthermore, our results support the S1-trapping hypothesis [J. J. Szymczak et al., J. Phys. Chem. A, 2009, 113, 12686–12693]. For thymine, the computational cost of a single TDDFT-SH time-step including the lowest 3 states, all couplings and gradients, is ∼5 times larger than the cost of a single Born–Oppenheimer dynamics time step for the ground state in our implementation. Thus, ps nonadiabatic dynamics simulations using multistate hybrid TDDFT-SH for systems with up to ∼100 atoms are possible without drastic approximations on single workstation nodes. Our implementation will be made available through Turbomole.
- This article is part of the themed collections: PCCP Editor’s Choice, 2020 and 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

