
A simple COSMO-based method for calculation of hydration energies of neutral molecules†
Alexander A.
Voityuk  *ab
and
Sergei F.
Vyboishchikov
*bc
*ab
and
Sergei F.
Vyboishchikov
*bc
*ab
and
Sergei F.
Vyboishchikov
*bc
Abstract
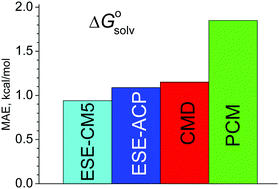
A simple, non-iterative method to estimate hydration free energies of neutral molecules, ESE, is developed. It requires only atomic charges computed for isolated species. To obtain the solvation free energy, the COSMO electrostatic term is supplemented by an extra correction that describes the cavitation energy, van der Waals and specific interactions. This term depends on atomic parameters that are adjusted using a reference dataset. Despite its simplicity, the ESE method provides accurate hydration energies with a mean absolute error below 1 kcal mol−1, superseding most accurate existing polarization continuum methods. We show that the proposed scheme can be directly extended to non-aqueous solutions.
 Please wait while we load your content...
Please wait while we load your content...

