
The water–carbon monoxide dimer: new infrared spectra, ab initio rovibrational energy level calculations, and an interesting in-termolecular mode†

Abstract
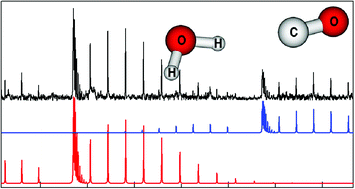
Bound state rovibrational energy level calculations using a high-level intermolecular potential surface are reported for H2O–CO and D2O–CO. They predict the ground K = 1 levels to lie about 20 (12) cm−1 above K = 0 for H2O–CO (D2O–CO) in good agreement with past experiments. But the first excited K = 1 levels are predicted to lie about 3 cm−1 below their K = 0 counterparts in both cases. Line strength calculations also indicate that mid-infrared transitions from the K = 0 ground state to this seemingly anomalous excited K = 1 state should be observable. These predictions are strikingly verified by new spectroscopic measurements covering the C–O stretch region around 2200 cm−1 for H2O–CO, D2O–CO, and HOD–CO, and the O–D stretch region around 2700 cm−1 for D2O–CO, HOD–CO, and DOH–CO. The experiments probe a pulsed supersonic slit jet expansion using tunable infrared quantum cascade laser or optical parametric oscillator sources. Discrete perturbations in the O–D stretch region give an experimental lower limit to the binding energy D0 of about 340 cm−1 for D2O–CO, as compared to our calculated value of 368 cm−1. Wavefunction plots are presented to help understand the intermolecular dynamics of H2O–CO. Coriolis interactions are invoked to explain the seemingly anomalous energies of the first excited K = 1 levels.
- This article is part of the themed collection: PCCP Editor’s Choice, 2020
 Please wait while we load your content...
Please wait while we load your content...

