
An accurate density functional theory for the vapor–liquid interface of chain molecules based on the statistical associating fluid theory for potentials of variable range for Mie chainlike fluids†

Abstract
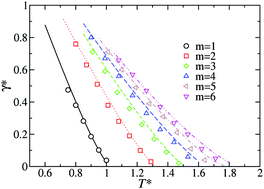
A new Helmholtz free energy density functional is presented to predict the vapor–liquid interface of chainlike molecules. The functional is based on the last version of the statistical associating fluid theory for potentials of variable range for homogeneous Mie chainlike fluids (SAFT-VR Mie). Following the standard formalism, the density functional theory (SAFT-VR Mie DFT) is constructed using a perturbative approach in which the free energy density contains a reference term to describe all the short-range interactions treated at the local level, and a perturbative contribution to account for the attractive perturbation which incorporates the long-range dispersive interactions. In this first work, we use a mean-field version of the theory in which the pair correlations are neglected in the attractive term. The SAFT-VR Mie DFT formalism is used to examine the effect of molecular chain length and the repulsive exponent of the intermolecular potential on density profiles and surface tension of linear chains made up of up to six Mie (λr − 6) segments with different values of the repulsive exponent of the intermolecular potential. Theoretical predictions from the theory are compared directly with molecular simulation data for density profiles and surface tension of Mie chainlike molecules taken from the literature. Agreement between theory and simulation data is good for short-chain molecules under all thermodynamic conditions of coexistence considered. Once the theory has proven that it is able to predict the interfacial properties, and particularly interfacial tension, the SAFT-VR Mie DFT formalism is used to predict the interfacial behavior of two new coarse-grained models for carbon dioxide and water recently proposed in the literature. In particular, the theoretical formalism, in combination with the coarse-grained models for carbon dioxide and water, is able to predict the interfacial properties of these important substances in a reasonable way.
 Please wait while we load your content...
Please wait while we load your content...

