
Anomalous kinetics of the reaction between OH and HO2 on an accurate triplet state potential energy surface

Abstract
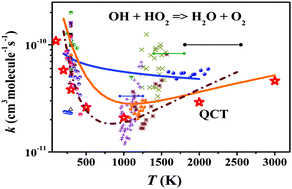
The reaction OH + HO2 → H2O + O2 is of great significance in interstellar media, the atmosphere, and combustion. In addition, it presents a prototypical reaction between two non-atom radical species. However, the temperature dependence of its rate coefficients has been debated for several decades. In this work, the rate coefficients are revisited by the quasi-classical trajectory (QCT) approach. To this end, a globally accurate full-dimensional potential energy surface of the ground triplet state for the title reaction is constructed using the permutation invariant polynomial-neural network (PIP-NN) method based on 108 000 points calculated at the level of CCSD(T)-F12a/AVTZ, in which particular attention is paid to the initial guess in the preceding Hartree–Fock procedure to obtain reliable ab initio energies. The QCT rate coefficients are compared to available experimental and theoretical results. It has been found that not only the trend, but also the magnitude, i.e. the large negative temperature dependence at low temperatures, and slightly positive temperature dependence at high temperatures, are consistent with some experiments.
 Please wait while we load your content...
Please wait while we load your content...

