
Ab initio computation for solid-state 31P NMR of inorganic phosphates: revisiting X-ray structures†
Kartik
Pilar,  a
Zeyu
Deng,
b
Molleigh B.
Preefer,
ac
Joya A.
Cooley,
a
Raphaële
Clément,
ad
Ram
Seshadri
acd
and
Anthony K.
Cheetham
*ae
a
Zeyu
Deng,
b
Molleigh B.
Preefer,
ac
Joya A.
Cooley,
a
Raphaële
Clément,
ad
Ram
Seshadri
acd
and
Anthony K.
Cheetham
*ae
a
Zeyu
Deng,
b
Molleigh B.
Preefer,
ac
Joya A.
Cooley,
a
Raphaële
Clément,
ad
Ram
Seshadri
acd
and
Anthony K.
Cheetham
*ae
Abstract
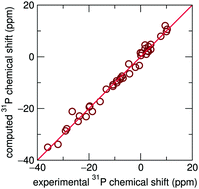
The complete 31P NMR chemical shift tensors for 22 inorganic phosphates obtained from ab initio computation are found to correspond closely to experimentally obtained parameters. Further improvement was found when structures determined by diffraction were geometry optimized. Besides aiding in spectral assignment, the cases where correspondence is significantly improved upon geometry optimization point to the crystal structures requiring correction.
- This article is part of the themed collection: 2024 Journal of Materials Chemistry Lectureship winner: Raphaële Clément
 Please wait while we load your content...
Please wait while we load your content...

