
Prediction of the near-IR spectra of ices by ab initio molecular dynamics

Abstract
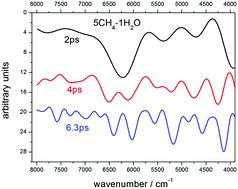
A method to predict the near-infrared spectra of amorphous solids by means of ab initio molecular dynamics is presented. These solids can simulate molecular ices. To test the method, mixtures of methane, water and nitrogen are generated as amorphous samples of various concentrations. The full theoretical treatment includes as a first step, the optimization of their geometrical structure for a range of densities, after which, the most stable systems are taken as initial structures for molecular dynamics, performed at 200 K in trajectories of 4 ps duration with a 0.2 fs time step. All the dynamics are carried out using the first principles method, solving the quantum problem for the electrons using density-functional theory (DFT), and integrating the DFT forces, following the Born–Oppenheimer dynamics. After the dynamics, near-IR spectra are predicted by the Fourier transform of the macroscopic polarization autocorrelation function. The calculated spectra are compared with the experimental spectra of ice mixtures of CH4 and H2O recorded in our laboratory, and with some spectra recorded by the New Horizons mission on Pluto.
 Please wait while we load your content...
Please wait while we load your content...

