
The influence of hydroxy groups on the adsorption of three-carbon alcohols on Ni(111), Pd(111) and Pt(111) surfaces: a density functional theory study within the D3 dispersion correction†

Abstract
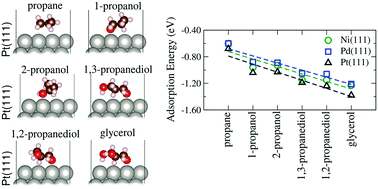
Experimentally, steric and inductive effects have been suggested as key parameters in the adsorption and reactivity of alcohols on transition-metal (TM) surfaces, however, our atomistic understanding of the behavior of alcohols in catalysis is far from satisfactory, in particular, due to the role of hydroxy groups in the adsorption properties of C3 alcohols on TM surfaces. In this study, we investigated those effects through ab initio calculations based on density functional theory employing a semilocal exchange–correlation functional within van der Waals corrections (the D3 framework) for the adsorption of C3 alcohols with different numbers and positions of OH groups, namely, propane, 1-propanol, 2-propanol, 1,2-propanediol, 1,3-propanediol and glycerol, on the compact Ni(111), Pd(111) and Pt(111) surfaces. As expected, we found that the adsorption energy is affected by the number of hydroxy groups with similar values for each pair of regioisomers, which clearly indicates the effect of the number of OH groups. Based on Bader charge analysis, we found an effective charge transfer from the C3 molecules to the substrates, which can explain the reduction in the work function due to adsorption. Upon adsorption, the alpha carbon to the OH group closest to the surface and the central carbon are the most positively charged atoms, which increases the lability of their bonded H atoms. In addition, the depletion of electron density between the C–H and O–H bonds closer to the surfaces corroborated their stretching, suggesting that the proximity of the adsorbates to the surfaces affects the acidity of these H atoms, as well as inductive effects within the molecules.
 Please wait while we load your content...
Please wait while we load your content...

