
Interactions between cyclic nucleotides and common cations: an ab initio molecular dynamics study†
 *a
Holger
Kruse
*a
and
*a
Holger
Kruse
*a
and
Abstract
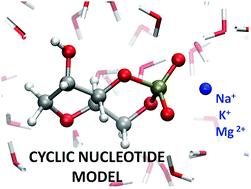
We present the first, to the best of our knowledge, ab initio molecular dynamics (AIMD) investigation on three aqueous solutions where an abasic cyclic nucleotide model is solvated in the presence of distinct cations (i.e., Na+, K+ and Mg2+). We elucidate the typical modalities of interaction between those ionic species and the nucleotide moiety by first-principles numerical simulations, starting from an inner-shell binding configuration on a time scale of 100 ps (total simulation time of ∼600 ps). Whereas the strong “structure-maker” Mg2+ is permanently bound to one of the two oxygen atoms of the phosphate group of the nucleotide model, Na+ and K+ show binding times τb of 65 ps and 10–15 ps, respectively, thus reflecting their chemical nature in aqueous solutions. Furthermore, we qualitatively relate these findings to approximate free-energy barriers of the cations' unbinding obtained by means of exploratory well-tempered metadynamics. With the aim of shedding light on the features of commonly employed force-fields (FFs), classical MD simulations (almost 200 trajectories with a total simulation time of ∼18 μs) using the biomolecular AMBER FF are also reported. By choosing several combinations of the parametrization for the water environment (i.e., TIP3P, SPC/E and OPC) and cations (i.e., Joung–Cheatham, Li–Merz 12-6 and Li–Merz 12-6-4), we found significant differences in the radial distribution functions and residence times compared to the ab initio results. The Na+ and K+ ions wrongly show quasi-identical radial distribution functions and the Li & Merz 12-6-4 Lennard-Jones parameters for Mg2+ were found to be essential in quickly reaching the binding state consistent with AIMD.
 Please wait while we load your content...
Please wait while we load your content...

