
Extremely large differences in DFT energies for nitrogenase models†

Abstract
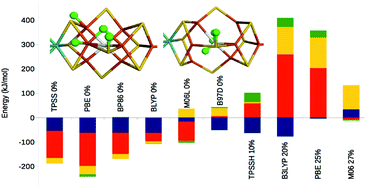
Nitrogenase is the only enzyme that can cleave the triple bond in N2, making nitrogen avaiable for other organisms. It contains a complicated MoFe7S9C(homocitrate) cluster in its active site. Many computational studies with density-functional theory (DFT) of the nitrogenase enzyme have been presented, but they do not show any consensus – they do not even agree where the first four protons should be added, forming the central intermediate E4. We show that the prime reason for this is that different DFT methods give relative energies that differ by almost 600 kJ mol−1 for different protonation states. This is 4–30 times more than what is observed for other systems. The reason for this is that in some structures, the hydrogens bind to sulfide or carbide ions as protons, whereas in other structures they bind to the metals as hydride ions, changing the oxidation state of the metals, as well as the Fe–C, Fe–S and Fe–Fe distances. The energies correlate with the amount of Hartree–Fock exchange in the method, indicating a variation in the amount of static correlation in the structures. It is currently unclear which DFT method gives the best results for nitrogenase. We show that non-hybrid DFT functionals and TPSSh give the most accurate structures of the resting active site, whereas B3LYP and PBE0 give the best H2 dissociation energies. However, no DFT method indicates that a structure of E4 with two bridging hydride ions is lowest in energy, as spectroscopic experiments indicate.
 Please wait while we load your content...
Please wait while we load your content...

