
Exploring the electrochemical properties of hole transporting materials from first-principles calculations: an efficient strategy to improve the performance of perovskite solar cells†
 a
Rongxing
He
*a
a
Rongxing
He
*a
Abstract
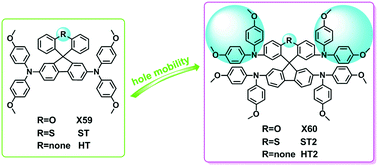
Perovskite solar cells (PSCs) have been achieved with impressively dynamic improvement in power conversion efficiency (PCE), becoming the hottest topic in photovoltaics. One of the hot topics is to develop inexpensive and efficient hole transporting materials (HTMs). In the present work, we systematically investigated the impact of different atoms in the heteromerous structure on the performance of perovskite solar cells. In addition, the influence of the structural modification of the HTM molecular building blocks was also revealed. To further understand the relationship between the charge-transport properties and the structural modification, the electronic properties, reorganization energy, and hole transporting properties of a series of organic hole transporting materials were investigated using first-principles calculations combined with Marcus theory. Moreover, the orientation function μΦ (V, λ, r, θ, γ; Φ) was applied to quantitatively evaluate the overall carrier mobility of HTMs in PSCs. It is revealed that this model predicts the hole mobility of HTMs correctly. The calculated results indicate that hole transporting materials with heteroatoms and larger dimensional structures show higher hole mobility, which may significantly improve the photovoltaic performance of PSCs.
 Please wait while we load your content...
Please wait while we load your content...

