
Ultrafast photodissociation dynamics of 2-ethylpyrrole: adding insight to experiment with ab initio multiple cloning†

Abstract
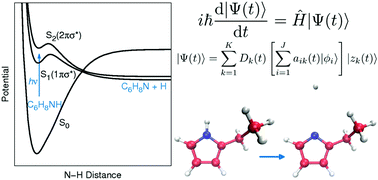
The ultrafast photodissociation dynamics of 2-ethylpyrrole (2-EP) is simulated in a fully quantum manner on the S1 and S2 πσ* states by the ab initio multiple cloning (AIMC) method. AIMC treats electrons with accurate electronic structure methods “on the fly”, and nuclear dynamics with wavefunction propagation via a basis set of Ehrenfest trajectory guided Gaussian wavepackets. Total kinetic energy release (TKER) spectra are produced, as well as velocity map images and N–H dissociation times. These are compared to results from time-resolved velocity map imaging studies, and the AIMC method is able to provide quantitative reproduction of experimental data, including dissociation times of 50–80 fs. Novel insight into the dissociation mechanism is then obtained, with the experimentally obtained time constant shown to be composed of two components. Firstly, there is a contribution in <50 fs from 2-EP molecules that have sufficient energy in the N–H stretch coordinate to dissociate almost immediately over the barrier, and this is followed by a second slower contribution from 2-EP molecules that must sample the potential energy surface before finding a way around the barrier to dissociate. This two component mechanism is not observed experimentally due to the temporal widths of the laser pulses obscuring the dynamics in the <50 fs window, and is shown for the first time via theory. Calculations are also performed on selectively deuterated 2-EP, demonstrating that AIMC is able to produce a kinetic isotope effect for the dissociation time constant, and correctly predict a shift to lower energy in the TKER spectrum. The S2 πσ* state is also shown to be unstable with respect to the S1 πσ* state, with the N–H dissociation proceeding along S1 when initially excited to S2. This work demonstrates that the combination of state of the art theory and experiments can provide unprecedented novel insight into the N–H dissociation mechanism, with the tantalising prospect of providing insight into more general heteroatom hydride bond dissociation.
 Please wait while we load your content...
Please wait while we load your content...

