
Defect formation in In2O3 and SnO2: a new atomistic approach based on accurate lattice energies
 *a
John
Buckeridge,
a
Tomas
Lazauskas,
a
David
Mora-Fonz,
b
Alexey A.
Sokol,
a
Scott M.
Woodley
a
and
*a
John
Buckeridge,
a
Tomas
Lazauskas,
a
David
Mora-Fonz,
b
Alexey A.
Sokol,
a
Scott M.
Woodley
a
and
Abstract
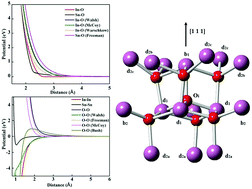
We present a consistent interatomic force field for indium sesquioxide (In2O3) and tin dioxide (SnO2) that has been derived to reproduce lattice energies and, consequently, the oxygen vacancy formation energies in the respective binary compounds. The new model predicts the dominance of Frenkel-type disorder in SnO2 and In2O3, in good agreement with ab initio defect calculations. The model is extended to include free electron and hole polarons, which compete with charged point defects to maintain charge neutrality in a defective crystal. The stability of electrons and instability of holes with respect to point defect formation rationalises the efficacy of n-type doping in tin doped indium oxide (ITO), a widely employed transparent conducting oxide in optoelectronic applications. We investigate the clustering of Sn substitutional and oxygen interstitial sites in ITO, finding that the dopants substitute preferentially on the cation crystallographic d site in the bixbyite unit cell, in agreement with experiment. The force field described here provides a useful avenue for the investigation of the defect properties of extended transparent conducting oxide systems, including solid solutions.
- This article is part of the themed collection: In memory of Paul O’Brien
 Please wait while we load your content...
Please wait while we load your content...

