
Systematic molecular model development with reliable charge distributions for gaseous adsorption in nanoporous materials†
 a
and
Li-Chiang
Lin
*a
a
and
Li-Chiang
Lin
*a
Abstract
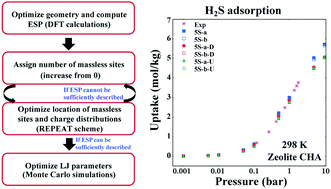
Adsorption technology using nanoporous materials has recently drawn considerable attention as a potentially energy-efficient approach for gas removal. To expedite the search for adsorbent candidates, molecular simulations play a critical role and their successful employment highly depends on the reliable description of the molecules. For this purpose, we develop a systematic and robust scheme for the model development of small gaseous molecules. This scheme enables an exhaustive and efficient search over all possible model parameters. By incorporating ab initio density functional theory calculations, the number and location of pseudo-sites as well as their atomic charge assignment can be efficiently determined with an accurate representation of the electrostatic potential (ESP) surface of the molecules. Subsequently, the van der Waals interaction parameters of the model are fitted to reproduce the experimental vapor–liquid equilibrium. This method has two major advantages: (1) accurate ESP surface descriptions of the developed models, and (2) significantly improved computational efficiency due to the decoupling of the force field parameters. As a proof of concept, we develop hydrogen sulfide (H2S) models, in the hope of using accurate H2S models to facilitate the discovery of potential adsorbents for its removal. It is found that an adequate H2S model should possess at least 2 massless sites, leading to models having five or more sites in order to describe the ESP with high accuracy. These models are demonstrated to well describe the adsorption phenomena of H2S in nanoporous materials, as well as to reproduce a variety of experimentally determined liquid properties. An extensive comparison to other existing H2S models is also carried out, where our models show substantial improvements. Furthermore, with the developed models, the adsorption properties of H2S in more than 160 siliceous zeolites are investigated, and a number of promising candidates are identified for H2S removal. The method and H2S models developed herein would pave the way for future studies on the discovery of cost-effective and environmentally friendly materials for numerous applications.
 Please wait while we load your content...
Please wait while we load your content...

