
Self-assembly of semiflexible polymers confined to thin spherical shells
 *a
and
Arash
Nikoubashman
a
*a
and
Arash
Nikoubashman
a
Abstract
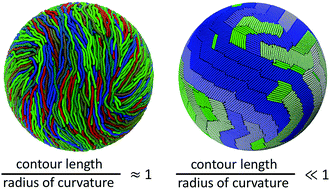
Confinement effects are critical for stiff macromolecules in biological cells, vesicles, and other systems in soft matter. For these molecules, the competition between the packing entropy and the enthalpic cost of bending is further shaped by strong confinement effects. Through coarse-grained molecular dynamics simulations, we explore the self-assembly of semiflexible polymers confined in thin spherical shells for various chain lengths, chain stiffnesses, and shell thicknesses. Here, we focus on the case where the contour and persistence length of the polymers are comparable to the radius of the confining cavity. The range of ordered structures is analyzed using several order parameters to elucidate the nature of orientational ordering in different parameter regimes. Previous simulations have revealed the emergence of bipolar and quadrupolar topological defects on the surface when the entire cavity was filled with a concentrated polymer solution [Phys. Rev. Lett., 2017, 118, 217803]. In contrast, spherical shell confinement restricts the appearance of a bipolar order. Instead, only the extent of the quadrupolar order changes with chain stiffness, as evidenced by the relative motion of topological defects. In the case of monolayers, we observe a nematic to smectic transition accompanied by a change in the nematic grain-size distribution as the contour length was decreased.
 Please wait while we load your content...
Please wait while we load your content...

