
Ab initio prediction of the polymorph phase diagram for crystalline methanol†
 a
and
Gregory J. O.
Beran
*b
a
and
Gregory J. O.
Beran
*b
Abstract
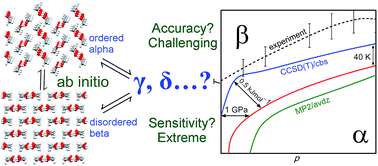
Organic crystals frequently adopt multiple distinct polymorphs exhibiting different properties. The ability to predict not only what crystal forms might occur, but under what experimental thermodynamic conditions those polymorphs are stable would be immensely valuable to the pharmaceutical industry and others. Starting only from knowledge of the experimental crystal structures, this study successfully predicts the methanol crystal polymorph phase diagram from first-principles quantum chemistry, mapping out the thermodynamic regions of stability for three polymorphs over the range 0–400 K and 0–6 GPa. The agreement between the predicted and experimental phase diagrams corresponds to predicting the relative polymorph free energies to within ∼0.5 kJ mol−1 accuracy, which is achieved by employing fragment-based second-order Møller–Plesset perturbation theory and coupled cluster theory plus a quasi-harmonic treatment of the phonons.
- This article is part of the themed collection: 2018 ChemSci Pick of the Week Collection
 Please wait while we load your content...
Please wait while we load your content...

