
Probing the oxidation state of transition metal complexes: a case study on how charge and spin densities determine Mn L-edge X-ray absorption energies†
 ‡a
Meiyuan
Guo,
‡§b
Michael L.
Baker,
e
Sheraz
Gul,
f
Jan
Kern,
fg
Alexander
Föhlisch,
ah
Uwe
Bergmann,
i
Junko
Yano,
f
Marcus
Lundberg
*b
and
Philippe
Wernet
*a
‡a
Meiyuan
Guo,
‡§b
Michael L.
Baker,
e
Sheraz
Gul,
f
Jan
Kern,
fg
Alexander
Föhlisch,
ah
Uwe
Bergmann,
i
Junko
Yano,
f
Marcus
Lundberg
*b
and
Philippe
Wernet
*a
Abstract
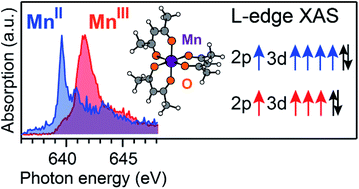
Transition metals in inorganic systems and metalloproteins can occur in different oxidation states, which makes them ideal redox-active catalysts. To gain a mechanistic understanding of the catalytic reactions, knowledge of the oxidation state of the active metals, ideally in operando, is therefore critical. L-edge X-ray absorption spectroscopy (XAS) is a powerful technique that is frequently used to infer the oxidation state via a distinct blue shift of L-edge absorption energies with increasing oxidation state. A unified description accounting for quantum-chemical notions whereupon oxidation does not occur locally on the metal but on the whole molecule and the basic understanding that L-edge XAS probes the electronic structure locally at the metal has been missing to date. Here we quantify how charge and spin densities change at the metal and throughout the molecule for both redox and core-excitation processes. We explain the origin of the L-edge XAS shift between the high-spin complexes MnII(acac)2 and MnIII(acac)3 as representative model systems and use ab initio theory to uncouple effects of oxidation-state changes from geometric effects. The shift reflects an increased electron affinity of MnIII in the core-excited states compared to the ground state due to a contraction of the Mn 3d shell upon core-excitation with accompanied changes in the classical Coulomb interactions. This new picture quantifies how the metal-centered core hole probes changes in formal oxidation state and encloses and substantiates earlier explanations. The approach is broadly applicable to mechanistic studies of redox-catalytic reactions in molecular systems where charge and spin localization/delocalization determine reaction pathways.
- This article is part of the themed collection: Editor's Choice – Serena DeBeer
 Please wait while we load your content...
Please wait while we load your content...

