
The mechanism of directed Ni(ii)-catalyzed C–H iodination with molecular iodine†

Abstract
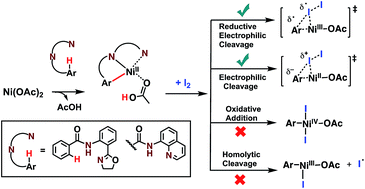
The density functional theory method is used to elucidate the elementary steps of Ni(II)-catalyzed C(sp2)–H iodination with I2 and substrates bearing N,N′-bidentate directing centers, amide-oxazoline (AO) and 8-aminoquinoline (AQ). The relative stability of the lowest energy high- and low-spin electronic states of the catalyst and intermediates is found to be an important factor for all of the steps in the reaction. As a result, two-state reactivity for these systems is reported, where the reaction is initiated on the triplet surface and generates a high energy singlet nickelacycle. It is shown that the addition of Na2CO3 base to the reaction mixture facilitates C–H activation. The presence of I2 in the reaction provides the much needed driving force for the C–H activation and nickelacycle formation and ultimately reacts to form a new C–I bond through either a redox neutral electrophilic cleavage (EC) pathway or a one-electron reductive cleavage (REC) pathway. The previously proposed Ni(II)/Ni(IV) and homolytic cleavage pathways are found to be higher in energy. The nature of the substrate is found to have a large impact on the relative stability of the lowest electronic states and on the stability of the nickelacycle resulting from C–H activation.
 Please wait while we load your content...
Please wait while we load your content...

