
Efficient prediction of reaction paths through molecular graph and reaction network analysis†

Abstract
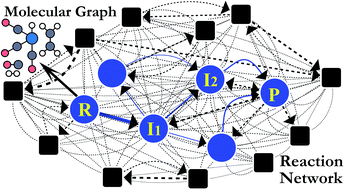
Despite remarkable advances in computational chemistry, prediction of reaction mechanisms is still challenging, because investigating all possible reaction pathways is computationally prohibitive due to the high complexity of chemical space. A feasible strategy for efficient prediction is to utilize chemical heuristics. Here, we propose a novel approach to rapidly search reaction paths in a fully automated fashion by combining chemical theory and heuristics. A key idea of our method is to extract a minimal reaction network composed of only favorable reaction pathways from the complex chemical space through molecular graph and reaction network analysis. This can be done very efficiently by exploring the routes connecting reactants and products with minimum dissociation and formation of bonds. Finally, the resulting minimal network is subjected to quantum chemical calculations to determine kinetically the most favorable reaction path at the predictable accuracy. As example studies, our method was able to successfully find the accepted mechanisms of Claisen ester condensation and cobalt-catalyzed hydroformylation reactions.
 Please wait while we load your content...
Please wait while we load your content...

