
Allotropes of tellurium from first-principles crystal structure prediction calculations under pressure

Abstract
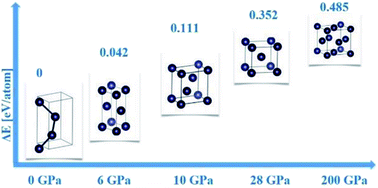
We investigated the allotropes of tellurium under hydrostatic pressure based on density functional theory calculations and crystal structure prediction methodology. Our calculated enthalpy-pressure and energy-volume curves unveil the transition sequence from the trigonal semiconducting phase, represented by the space group P3121 in the range of 0–6 GPa, to the body centered cubic structure, space group Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, stable at 28 GPa. In between, the calculations suggest a monoclinic structure, represented by the space group C2/m and stable at 6 GPa, and the β-Po type structure, space group Rm, stable at 10 GPa. The face-centered structure is found at pressure as high as 200 GPa. As the pressure is increased, the transition from the semiconducting phase to metallic phases is observed.
m, stable at 28 GPa. In between, the calculations suggest a monoclinic structure, represented by the space group C2/m and stable at 6 GPa, and the β-Po type structure, space group Rm, stable at 10 GPa. The face-centered structure is found at pressure as high as 200 GPa. As the pressure is increased, the transition from the semiconducting phase to metallic phases is observed.
 Please wait while we load your content...
Please wait while we load your content...

