
Halogen bonding for the design of inhibitors by targeting the S1 pocket of serine proteases†

Abstract
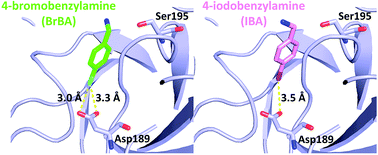
Halogen bonding (or X bonding) has attracted increasing interest due to its significant role in molecular recognition in biological systems. Trypsin-like serine proteases have many physiological and pathophysiological functions. There is therefore extensive interest in generating specific inhibitors for pharmacological intervention in their enzymatic activity. We study here if it is possible to use halogenated compounds as the P1 group to bind to the S1 specificity pocket of trypsin-like serine proteases to avoid the low bioavailability of the amidine or guanidine P1 group that is typically used in many inhibitors. We used 4-chlorobenzylamine (ClBA), 4-bromobenzylamine (BrBA) and 4-iodobenzylamine (IBA) as probes to test their binding modes to a trypsin-like serine protease, urokinase-type plasminogen activator (uPA), which has been recognized as a marker for breast cancer and an important target for inhibitor development. The results showed that these compounds inhibited uPA with stronger efficacies compared with their non-halogenated analogues. We also determined the high-resolution crystal structures of uPA in complex with BrBA and IBA, respectively. The structures revealed that BrBA bound to the S1 pocket of uPA via halogen bonds, but IBA did not make halogen bonds with uPA, demonstrating that the iodine may not be the best choice as a target moiety for serine proteases. These results advocate halogen bonding, especially bromine bonding, as an efficient strategy for the future design of novel inhibitors against trypsin-like serine proteases to provide strong potency and promote bioavailability.
- This article is part of the themed collection: Editors' collection: Chemical Biology
 Please wait while we load your content...
Please wait while we load your content...

