
The effect of the intramolecular C–H⋯O interactions on the conformational preferences of bis-arylsulfones – 5-HT6 receptor antagonists and beyond†

Abstract
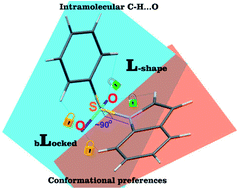
The development of compounds with enhanced activity and selectivity by a conserved spatial orientation of the pharmacophore elements has a long history in medicinal chemistry. Rigidified compounds are an example of this concept. However, the intramolecular interactions were seldom used as a basis for conformational restraints. Here, we show the weak intramolecular interactions that contribute to the relatively well-conserved geometry of N1-arylsulfonyl indole derivatives. The structure analysis along with quantum mechanics calculations revealed a crucial impact of the sulfonyl group on the compound geometry. The weak intramolecular C–H⋯O interaction stabilizes the mutual "facing" orientation of two aromatic fragments. These findings extend the pharmacological interpretation of the sulfonyl group role from the double hydrogen bond acceptor to the conformational scaffold based on intramolecular forces. This feature has, to date, been omitted in in silico drug discovery. Our results should increase the awareness of researchers to consider the conformational preference when designing new compounds or improving computational methods.
 Please wait while we load your content...
Please wait while we load your content...

