
BxCyNz hybrid graphenylene: stability and electronic properties†

Abstract
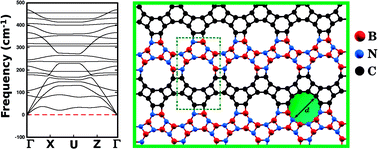
Interest in hybrid monolayers with arrangements that differ from that of the honeycomb lattice has been growing. However, systematic investigations on the properties of these structures are still lacking. In this work, we combined density functional theory (DFT) and molecular dynamics (MD) simulations to study the stability and electronic properties of nanosheets composed of B, C, and N atoms arranged in the pattern of the carbon allotrope graphenylene. We considered twenty structures with varied atomic arrangements and stoichiometries, which we call BxCyNz hybrid graphenylenes. We calculated the formation energy for each arrangement, and found that it decreases as the number of B–C and N–C bonds decreases. We also found that the structure with minimum energy has stoichiometry B2CN and an atomic arrangement with BN and C stripes connected along the zigzag direction. Regarding the electronic properties, we found that all investigated structures are semiconductors, with band gaps ranging from 0.14 to 1.65 eV. Finally, we found that the optimized hybrid lattices presented pores of varied sizes and shapes. This diversity in pore geometry suggests that these structures might be particularly suited for molecular sieve applications.
 Please wait while we load your content...
Please wait while we load your content...

