
Electronic properties of the coronene series from thermally-assisted-occupation density functional theory†

Abstract
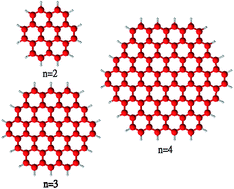
To fully utilize the great potential of graphene in electronics, a comprehensive understanding of the electronic properties of finite-size graphene flakes is essential. While the coronene series with n fused benzene rings at each side (designated as n-coronenes) are possible structures for opening a band gap in graphene, their electronic properties are not yet fully understood. Nevertheless, because of their radical character, it remains very difficult to reliably predict the electronic properties of the larger n-coronenes with conventional computational approaches. In order to circumvent this, the various electronic properties of n-coronenes (n = 2–11) are investigated using thermally-assisted-occupation density functional theory (TAO-DFT) [J.-D. Chai, J. Chem. Phys., 2012, 136, 154104], a very efficient electronic structure method for studying nanoscale systems with strong static correlation effects. The ground states of the larger n-coronenes are shown to be polyradical singlets, where the active orbitals are mainly localized at the zigzag edges.
 Please wait while we load your content...
Please wait while we load your content...

