
Mechanism investigation on the reactions of ClF3O and n-decane by combining density functional theory and spontaneous emission spectroscopy†
 a
a
Abstract
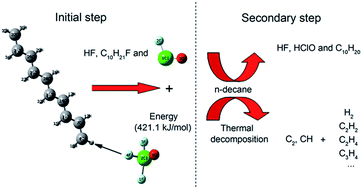
The mechanism of the reactions of ClF3O and n-decane had two stages. The first stage was the initial reaction between ClF3O and n-decane. The initial reactions were investigated using a density functional theory (DFT) method. The results showed that the critical part of the mechanism of the initial reaction was the roaming of the HF intermediate. A H atom on n-decane was abstracted by a F atom on ClF3O to produce HF. The formed HF roamed around and then broke to give ClFO, fluorinated decane and a new HF molecule. The initial reactions were considered to be barrier-less reactions and extremely exothermic. The average released energy of the initial reactions was 412.9 kJ mol−1, which was great enough to cause thermal decomposition of n-decane. The second stage included the reaction between ClFO and n-decane and the thermal decomposition of n-decane. The secondary reactions involving ClFO were also studied using a DFT method. ClFO was less reactive than ClF3O. The average energy barrier of the reactions of ClFO and n-decane was 116.3 kJ mol−1 and the average released energy was 266.5 kJ mol−1. Thermal decomposition of n-decane was evidenced by the emission spectra of the characteristic radical intermediates CH and C2, which were observed using an intensified charge-coupled device (ICCD) system. The main gaseous products of the thermal decomposition of n-decane, as identified using gas chromatography, were hydrogen, ethylene and acetylene. The experimental results showed that the thermal decomposition of n-decane was an important secondary reaction following the initial reactions.
 Please wait while we load your content...
Please wait while we load your content...

